Method
Determinig the orientation of the protein to the membrane bilayer
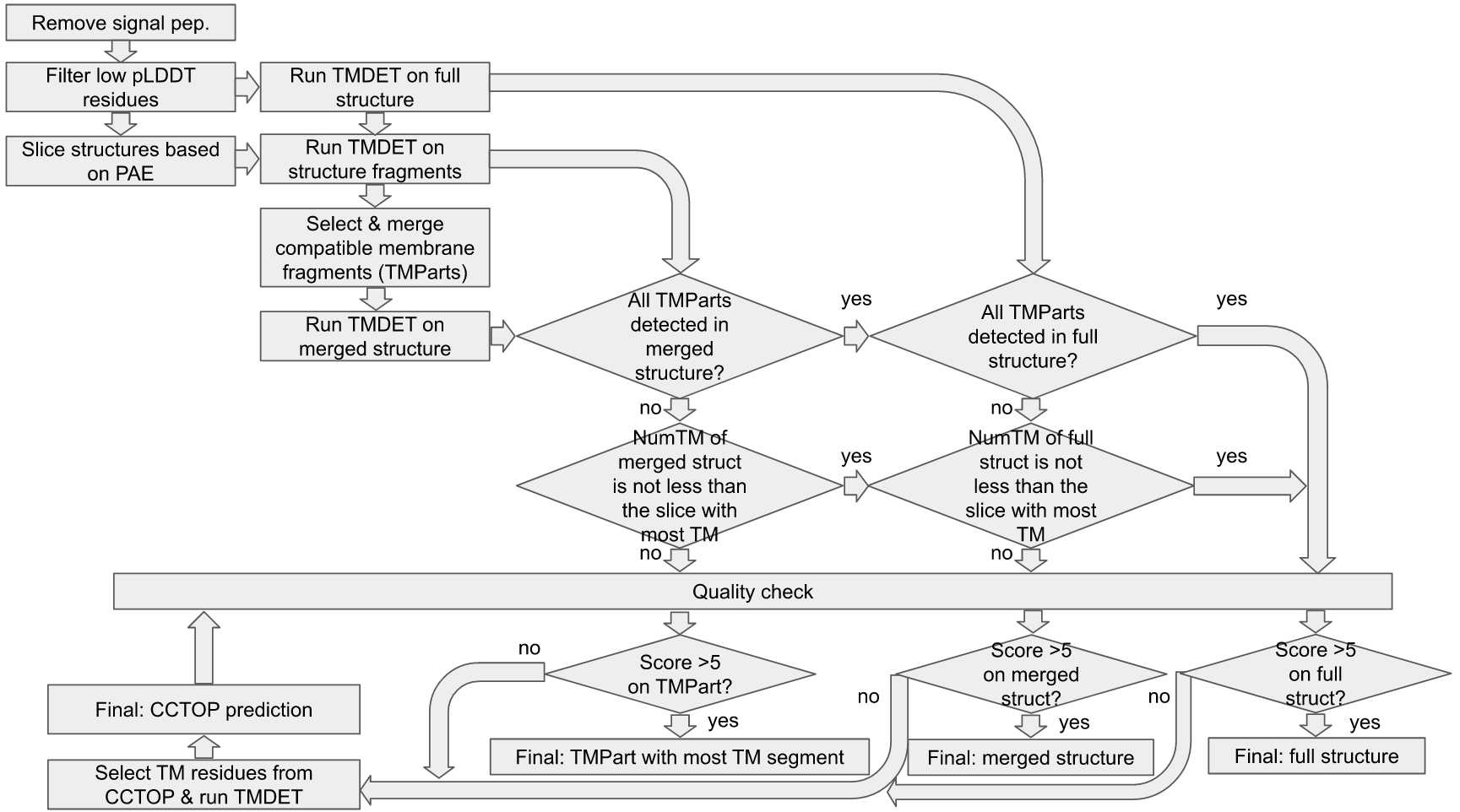 Algorithm's summary: In the TmAlphaFold database, we reconstruct the membrane bilayer using the PDB and Pairwise Alignment Error files provided by AlphaFold database. Briefly, we slice the full structure into potential domains using Agglomerative Clustering, then we use the TMDET algorithm to detect the membrane bilayer on the structure file(s). If the result is not satisfying (i.e. the final evaluation is Fail, see below) we also exploit CCTOP to find membrane segments and rerun TMDET only on the predicted transmembrane (TM) helices.
Algorithm's summary: In the TmAlphaFold database, we reconstruct the membrane bilayer using the PDB and Pairwise Alignment Error files provided by AlphaFold database. Briefly, we slice the full structure into potential domains using Agglomerative Clustering, then we use the TMDET algorithm to detect the membrane bilayer on the structure file(s). If the result is not satisfying (i.e. the final evaluation is Fail, see below) we also exploit CCTOP to find membrane segments and rerun TMDET only on the predicted transmembrane (TM) helices.
Algorithm in details:
- First, we remove the signal peptide predicted by SignalP6.
- After that, we mask residues based on DSSP secondary structure and pLDDT values: everything below 50 pLDDT are masked out (i.e. these residues were not taken into account by TMDET), helical residues are allowed between 50 and 70 pLDDT for the further calculations and any other residues above 70 pLDDT.
- Next we use the Pairwise Alignment Error (PAE) matrix to divide the structure into fragments, similarly as it is used in Chimera: PAE gives the expected position error at residue x, when the predicted and true structures are aligned on residue y. This matrix can be used to detect domains, or sometimes segments where the position of amino acids relative to each other is certain. For this task, we use Agglomerative Clustering with a threshold set to 7.
- Next, we run the TMDET algorithm on each fragment: it will rotate each structure so their Z coordinate will be perpendicular to the membrane plane (if it can be detected). Fragments, where the membrane bilayer can be constructed, will be checked whether they are rotated roughly at the same degree (with 15° flexibility) - then compatible fragments with the most TM segment will be merged into one structure. Meanwhile, we also run TMDET on the full structure (except for masked residues).
- The results from different TMDET runs are then compared to each other, where we aim to select the structure using the following conditions: I) all TM helices from the fragments are detected in the selected structure II) the least number of residues is masked/omitted III) structure with most TM helix is selected. After this process either the full structure, the merged structure (from compatible fragments) or the fragment alone is selected.
- The next part is to detect potential errors in the structure (see details in Quality check). If the selected structure passes more than 5 criteria, it will be selected and the process ends. In case of a lower score, we suppose the membrane plane cannot be determined using the PDB and PAE files alone, and we will use the CCTOP to predict TM segments. Predicted TM residues (± 5 amino acids) are kept and everything else were masked out. We run TMDET on the resulted structure and repeat the Quality check to define potential errors.
Examples how we select the final structure in TmAlphaFold database
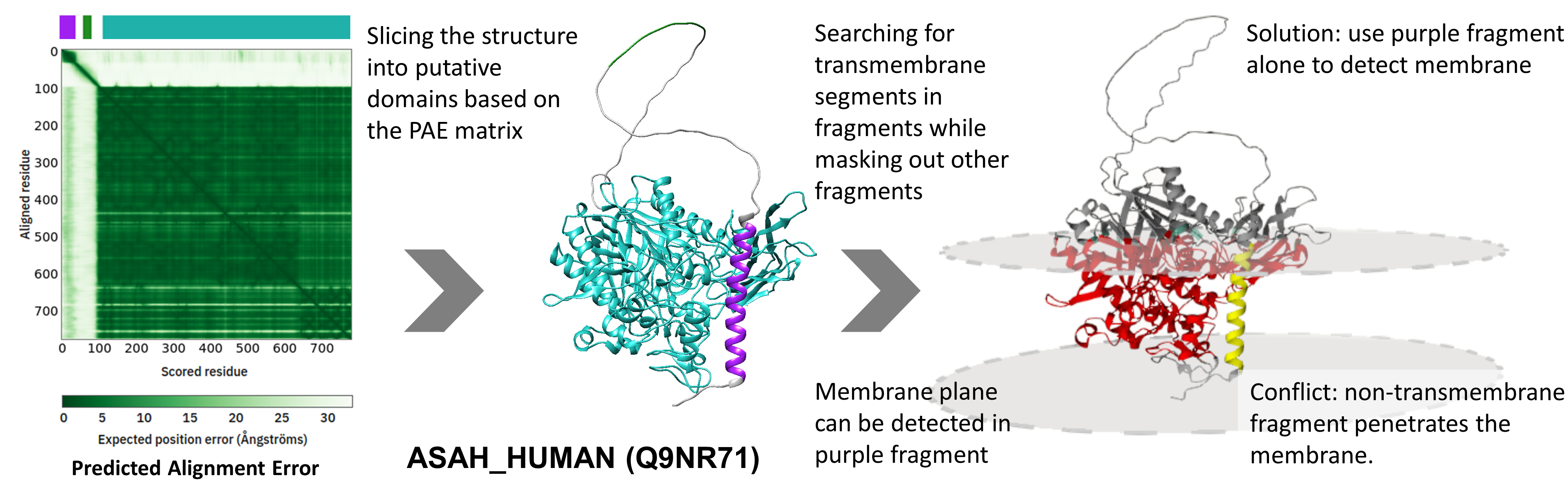 In most of the cases, the full structure can be used to detect the membrane plane after some consideration (i.e. removing signal peptides and low reliability residues). In other cases, slicing the structure using the Pairwise Alignment Error matrix to generate fragments can eliminate errors..
In most of the cases, the full structure can be used to detect the membrane plane after some consideration (i.e. removing signal peptides and low reliability residues). In other cases, slicing the structure using the Pairwise Alignment Error matrix to generate fragments can eliminate errors..ASAH2_HUMAN (Q9NR71) is a bitopic membrane protein, where a flexible segment connects the ceramidase domain with the TM segment. The flexible linker however turns and places the domain into the membrane plane, making it impossible to find the membrane plane. After slicing the full structure (colored on the figure), the TM helix can be distinguished and the membrane plane can be correctly detected.
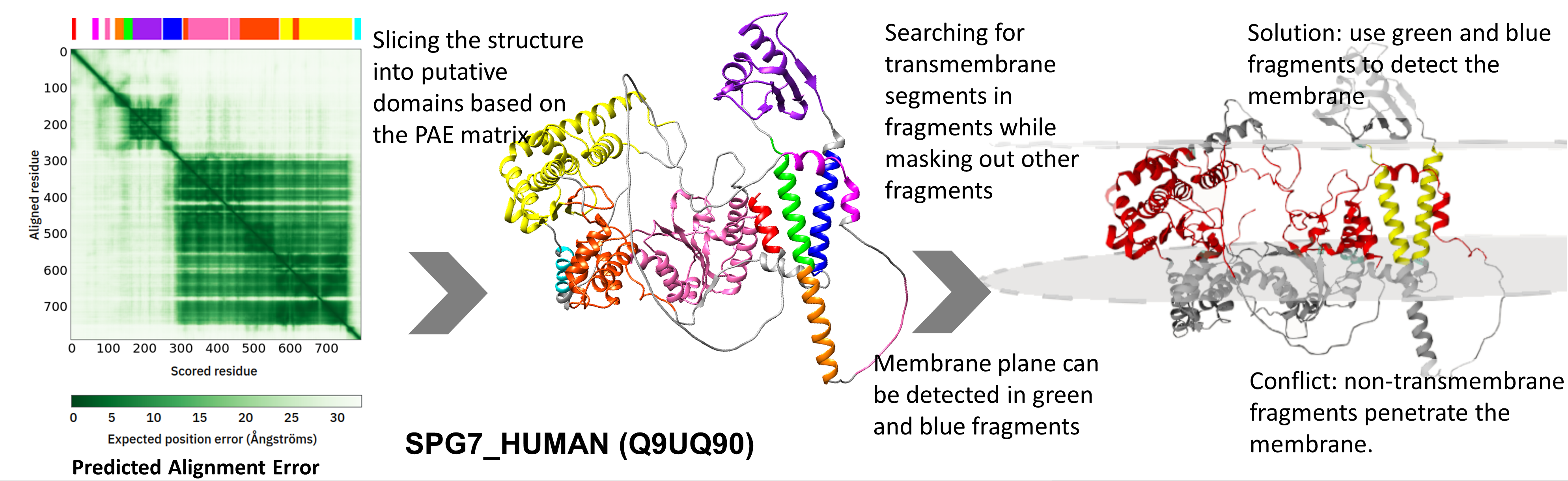 A more elaborate example is SPG7_HUMAN (Q9UQ90), a mitochondrion inner membrane protein where the membrane plane can be detected in two fragments. Structure parts with TM segments are checked for compatibility (i.e. if their transformation rotated them to roughly at the same degree) and the ones that agree are combined, so the merged structure will be used to detect the membrane plane.
A more elaborate example is SPG7_HUMAN (Q9UQ90), a mitochondrion inner membrane protein where the membrane plane can be detected in two fragments. Structure parts with TM segments are checked for compatibility (i.e. if their transformation rotated them to roughly at the same degree) and the ones that agree are combined, so the merged structure will be used to detect the membrane plane.
 PTH2R_HUMAN (P49190) is a GPCR-superfamily hormone receptor with 7 transmembrane helices. The protein also has an N-terminal signal peptide that is cleaved in the mature protein, although AF2 folds this stretch into the membrane domain. The C-terminal segment is predicted to be disordered , however, this region is also (erroneously) positioned inside the proposed membrane layer according to AF2. By filtering these segments out, the correct membrane bilayer can be reconstructed.
PTH2R_HUMAN (P49190) is a GPCR-superfamily hormone receptor with 7 transmembrane helices. The protein also has an N-terminal signal peptide that is cleaved in the mature protein, although AF2 folds this stretch into the membrane domain. The C-terminal segment is predicted to be disordered , however, this region is also (erroneously) positioned inside the proposed membrane layer according to AF2. By filtering these segments out, the correct membrane bilayer can be reconstructed.
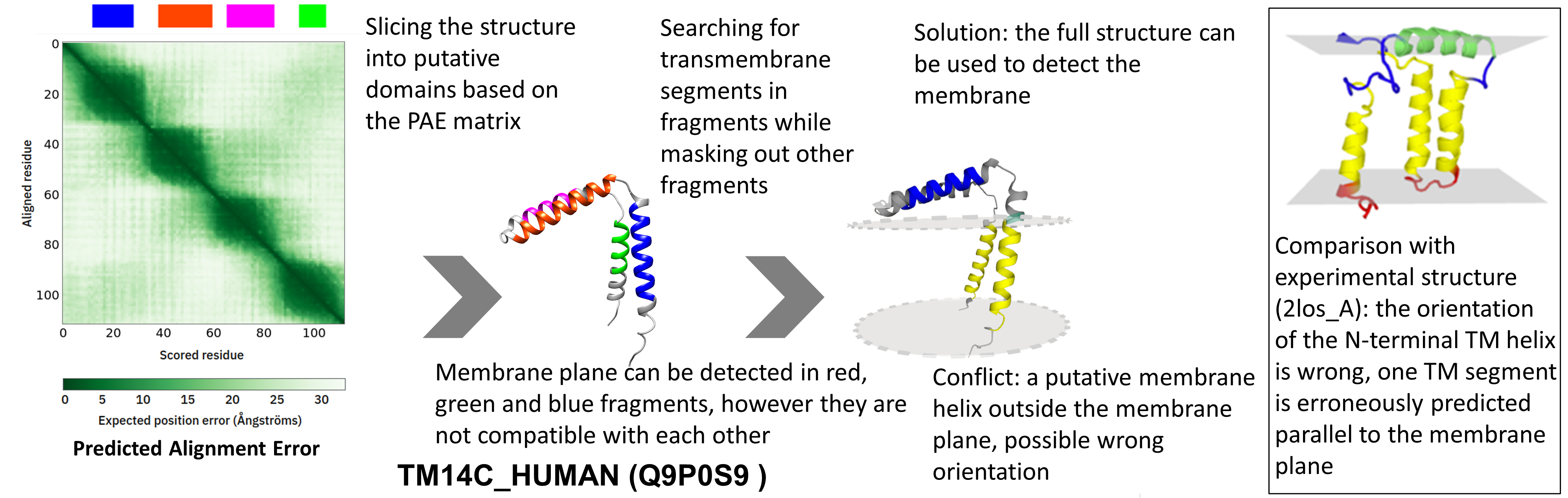 TM14C_HUMAN (Q9P0S9) is a transmembrane protein, probably involved in heme biosynthesis. AF2 predicts 4 alpha-helix in the protein, 2 pairs from which both could define the membrane plane. Considering the NMR structure of the protein, AF2 correctly identifies all secondary structure, however their orientation relative to each other is wrong – the middle TM helix is predicted parallel to the membrane next to the interface helix, and therefore the orientation of the N-terminal TM helix compared to the C-terminal one is wrong. NMR structures were not used to train AF2, yet it was shown that DeepMind’s algorithm is still accurate on them - with this exception that proves the rule.
TM14C_HUMAN (Q9P0S9) is a transmembrane protein, probably involved in heme biosynthesis. AF2 predicts 4 alpha-helix in the protein, 2 pairs from which both could define the membrane plane. Considering the NMR structure of the protein, AF2 correctly identifies all secondary structure, however their orientation relative to each other is wrong – the middle TM helix is predicted parallel to the membrane next to the interface helix, and therefore the orientation of the N-terminal TM helix compared to the C-terminal one is wrong. NMR structures were not used to train AF2, yet it was shown that DeepMind’s algorithm is still accurate on them - with this exception that proves the rule.
 AlphaFold2 aims to produce compact structures, that may lead folding additional elements to the membrane domain. For example OR1L1_HUMAN (Q8NH94) is an olfactory receptor, with seven TM region. AF2 places an extra helical segment next to the transmembrane segments. Since there is no geometrical violation, this is not detected during the quality check as a problem. Notably, according to UniProt it is uncertain that the initiating Methionin can be found at position 1 or 51 – in the latter case AF2 would probably predict a correct structure. TmAlphaFold Database may also fail on erroneous structures – sometimes AlphaFold2 wraps other segments of the protein around the TM helix or fails to assign the correct secondary structure for the TM segment.
AlphaFold2 aims to produce compact structures, that may lead folding additional elements to the membrane domain. For example OR1L1_HUMAN (Q8NH94) is an olfactory receptor, with seven TM region. AF2 places an extra helical segment next to the transmembrane segments. Since there is no geometrical violation, this is not detected during the quality check as a problem. Notably, according to UniProt it is uncertain that the initiating Methionin can be found at position 1 or 51 – in the latter case AF2 would probably predict a correct structure. TmAlphaFold Database may also fail on erroneous structures – sometimes AlphaFold2 wraps other segments of the protein around the TM helix or fails to assign the correct secondary structure for the TM segment.
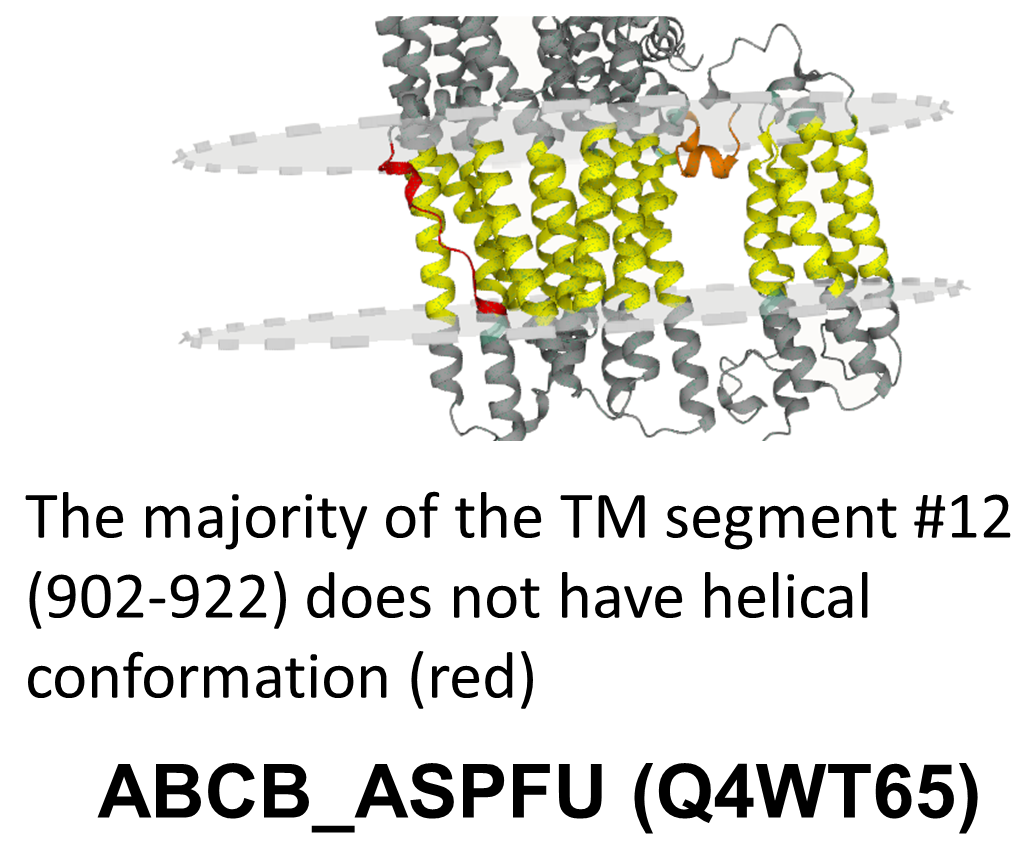 ABCB_ASPFU (Q4WT65) is an ATP-binding cassette transporter, providing an example where most of the TM domain is folded correctly, except one TM helix (902-922) - according to the Predicted Alignment Error matrix this region could not be reliabily aligned to the rest of the TM domain – our fragmentation also handles it as a separate entity.
ABCB_ASPFU (Q4WT65) is an ATP-binding cassette transporter, providing an example where most of the TM domain is folded correctly, except one TM helix (902-922) - according to the Predicted Alignment Error matrix this region could not be reliabily aligned to the rest of the TM domain – our fragmentation also handles it as a separate entity.
Quality check
In the TmAlphaFold database we display the following type of errors in the predicted structure
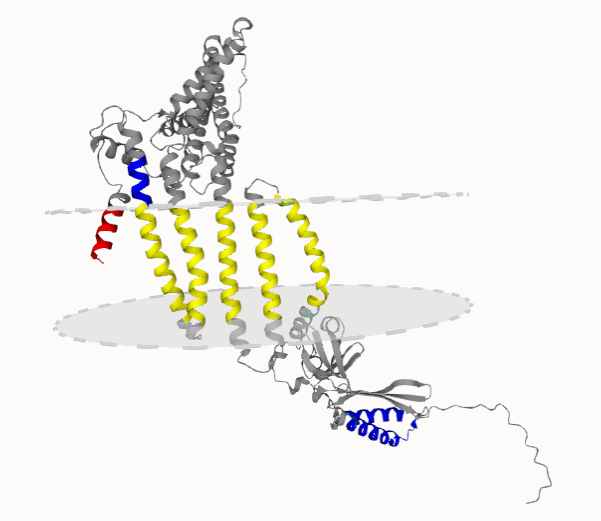 These errors are visible on the 3D Tab of the protein page and the structure is also colored in accordance. TM helices that seem to be correct are highlighted with yellow color. Segments in the membrane plane that are not supposed to be there are marked with red (these errors can raise from Short helix, Masked, Masked cross, Domain, Domain cross and Underpredict cctop). In contrast, segments expected to harbor the membrane, yet placed outside the membrane plane are blue. For more details see the “How to use the TmAlphaFold database” page.
These errors are visible on the 3D Tab of the protein page and the structure is also colored in accordance. TM helices that seem to be correct are highlighted with yellow color. Segments in the membrane plane that are not supposed to be there are marked with red (these errors can raise from Short helix, Masked, Masked cross, Domain, Domain cross and Underpredict cctop). In contrast, segments expected to harbor the membrane, yet placed outside the membrane plane are blue. For more details see the “How to use the TmAlphaFold database” page.
- Detecting membrane plane
- Returns passed if the membrane plane can be detected and constructed based on the AlphaFold structure.
- Signal
- AlphaFoldDB does not remove cleavable protein segments, and signal peptides may be embedded in the membrane plane erroneously. Returns passed if there is no signal peptide or if it is not embedded in the membrane.
- Full structure
- AlphaFold supplies a Pairwise Alignment Error, that gives the expected position error at residue x, when the predicted and true structures are aligned on residue y. This matrix can be used to detect domains in the structure. To reconstruct the membrane plane, TmAlphaFold cuts the structures to several pieces to detect the membrane, then reassembles them. Sometimes some of these parts are not compatible with the detected membrane plane - in such case TmAlphFold database still provides the membrane plane, however incompatible segments are masked out. Returns passed if all fragments were used to define the membrane plane.
- Short helix
- AlphaFold sometimes folds short helices to the membrane domain, or in other cases a longer helix partially gets masked out because of very low (<50) pLDDT scores. These segments are still detected, however the protein gets passed flag, if all transmembrane helix is longer than 10 amino acids.
- Masked segment in membrane plane
- AlphaFold provides reliability for predicted each residue in the structure. Residues under 50 pLDDT or non-helical residues under 70 pLDDT are masked out when the membrane plane is reconstructed. When the membrane plane is defined, TmAlphFold checks if masked regions are embedded in the membrane. Returns passed if there is no such region.
- Missing transmembrane part
- TmAlphFold cuts the structures to several pieces to detect the membrane then assembles the compatible pieces define the final membrane plane. In some cases the membrane plane can be only detected if several pieces are left out - even if a (non-compatible) membrane can be detected in them. Return passed if the assembly and the fragments are consistent.
- Domain or beta structure in membrane plane
- After splitting the structure, in some pieces (that are likely globular domains) no membrane will be detected. After reassembling the full structure, such parts should not embed into the membrane. Returns passed if no non-transmembrane piece interfere with the membrane plane or beta secondary structure appears.
- Overpredict cctop
- Comparing the result to the CCTOP topology prediction. Based on the membrane plane, membrane segments can be defined. Returns passed if there is no extra transmembrane segment in the TmAlphFold database structure.
- Underpredict cctop
- After defining membrane segments using the the membrane plane, returns passed if there is no extra transmembrane segment in the CCTOP prediction compared to the TmAlphFold database structure.
- Membrane plane cctop
- Even if there are extra/missing transmembrane segments, we can consider the membrane plane correctly reconstructed, if some of them overlap (thus the membrane plane can be found, however some parts of the polypeptid chain are predicted erroneously). Returns passed I) for bitopic membrane proteins if the transmembrane segment was found and II) for polytopic membrane proteins if at least two of them were found.
These errors are visible on the 3D Tab of the protein page and the structure is also colored in accordance. TM helices that seem to be correct are highlighted with yellow color. Segments in the membrane plane that are not supposed to be there are marked with red (these errors can raise from Short helix, Masked, Masked cross, Domain, Domain cross and Underpredict cctop). In contrast, segments expected to harbor the membrane, yet placed outside the membrane plane are blue. For more details see the “How to use the TmAlphaFold database” page.
After collecting all quality checks, a final evaluaion is given to every structure, simply by summing up the “passed” flags. Based on the score we categorize each protein as:
- Excellent (10)
- Good (9)
- Fair (6-8)
- Poor (5)
- Failed (0-4)
By using TmAlphaFold database you accept the Privacy Notice in compliance with Europe’s new General Data Protection Regulation (GDPR) that applies since 25 May 2018. Read the Privacy Notice